









Съдържание
Туберозна склероза на Борневил
Какво е ?
Туберозната склероза на Бурневил е сложно генетично заболяване, характеризиращо се с развитие на доброкачествен (нераков) тумор в различни части на тялото. След това тези тумори могат да бъдат локализирани в кожата, мозъка, бъбреците и други органи и тъкани. Тази патология също може да причини сериозни проблеми в развитието на индивида. Въпреки това, клиничните прояви и тежестта на заболяването варират при всеки пациент.
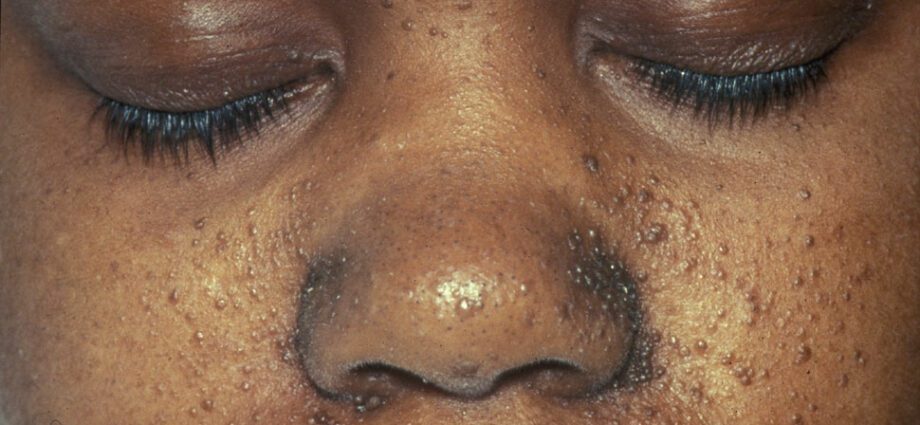
Свързаните кожни аномалии обикновено са подобни на петна по кожата или на области, където кожата е по-светла, отколкото на останалата част от тялото. Развитието на тумори в лицето се нарича ангиофиброма.
В контекста на мозъчно увреждане клиничните признаци са епилептични припадъци, поведенчески проблеми (хиперактивност, агресивност, интелектуални затруднения, проблеми с обучението и др.). Някои деца с това заболяване дори имат някаква форма на аутизъм, нарушения в развитието, засягащи социалните взаимодействия и комуникацията. Доброкачествените мозъчни тумори също могат да причинят усложнения, които могат да бъдат фатални за субекта.
Развитието на тумори в бъбреците е често срещано при хора с туберозна склероза. Това може да причини тежки усложнения при бъбречната функция. В допълнение, тумори могат да се развият в сърцето, белите дробове и ретината. (2)
Това е рядко заболяване, чието разпространение (брой на случаите в дадена популация в даден момент) възлиза на 1 / 8 до 000 / 1 души. (15)
Симптоми
Клиничните прояви, свързани с туберозната склероза на Bourneville, варират в зависимост от засегнатите органи. Освен това симптомите, свързани с болестта, варират в широки граници от един човек до друг. Със симптоми, вариращи от леки до тежки.
Най-широко идентифицираните симптоми на това заболяване включват епилептични припадъци, когнитивни и поведенчески разстройства, кожни аномалии и др. Най-често засегнатите органи са: мозъка, сърцето, бъбреците, белите дробове и кожата.
При това заболяване е възможно развитието на злокачествени (ракови) тумори, но са редки и засягат предимно бъбреците.
Клиничните признаци на заболяването в мозъка произлизат от атаки на различни нива:
– увреждане на кортикалните туберкули;
– епендимни възли (SEN);
– гигантски епендимни астроцитоми.
Те водят до: развитие на умствена изостаналост, обучителни затруднения, поведенчески разстройства, агресивност, разстройство на вниманието, хиперактивност, обсесивно-компулсивни разстройства и др.
Увреждането на бъбреците се характеризира с развитие на кисти или ангиомиолипоми. Те могат да доведат до бъбречна болка и дори до бъбречна недостатъчност. Ако се забелязва силно кървене, то може да е от тежка анемия или високо кръвно налягане. Други по-сериозни, но редки последици също могат да бъдат видими, по-специално развитие на карциноми (тумор на съставните клетки на епитела).
Увреждането на очите може да бъде подобно на видимите петна по ретината, причинявайки зрителни нарушения или дори слепота.
Кожните аномалии са многобройни:
– хипомеланични макули: които водят до появата на светли петна по кожата, навсякъде по тялото, в резултат на дефицит на меланин, протеин, който придава цвят на кожата;
– появата на червени петна по лицето;
– обезцветени петна по челото;
– други кожни аномалии, зависещи от един индивид на друг.
Белодробни лезии са налице при 1/3 от пациентите с лек превес при жените. Свързаните симптоми тогава са повече или по-малко тежки затруднения в дишането.
Произходът на болестта
Произходът на заболяването е генетичен и наследствен.
Предаването включва мутации в гените TSC1 и TSC2. Тези интересни гени влизат в игра при образуването на протеини: хамартин и туберин. Тези два протеина правят възможно чрез интерактивна игра да се регулира клетъчната пролиферация.
Пациентите с болестта се раждат с поне едно мутирало копие на тези гени във всяка от техните клетки. След това тези мутации ограничават образуването на хамартин или тубертин.
В контекста, където двете копия на гена са мутирали, те напълно предотвратяват производството на тези два протеина. Следователно този протеинов дефицит вече не позволява на тялото да регулира растежа на определени клетки и в този смисъл води до развитието на туморни клетки в различни тъкани и/или органи.
Рискови фактори
Рисковите фактори за развитие на такава патология са генетични.
Всъщност предаването на болестта е ефективно чрез автозомно доминантен начин. Или, мутиралият ген, който представлява интерес, се намира в несексуална хромозома. Освен това наличието само на едно от двете копия на мутиралия ген е достатъчно за развитието на болестта.
В този смисъл индивид, притежаващ един от тези двама родители, страдащи от заболяването, има 50% риск сам да развие болния фенотип.
Превенция и лечение
Диагнозата на заболяването е преди всичко диференциална. Тя се основава на нетипични физически критерии. В повечето случаи първите характерни признаци на заболяването са: наличие на повтарящи се епилептични припадъци и забавяне в развитието на субекта. В други случаи тези първи признаци водят до кожни петна или идентифициране на сърдечен тумор.
След тази първа диагноза са необходими допълнителни прегледи, за да се потвърди диагнозата или не. Те включват:
– сканиране на мозъка;
– ЯМР (магнитен резонанс) на мозъка;
– ултразвук на сърцето, черния дроб и бъбреците.
Диагнозата може да бъде ефективна при раждането на детето. В противен случай е важно то да се извърши възможно най-бързо, за да се поеме грижата за пациента възможно най-скоро.
В момента няма лек за болестта. Следователно свързаните лечения са независими от симптомите, представени от всеки индивид.
Обикновено се дават антиепилептични лекарства за ограничаване на припадъците. Освен това се предписват и лекарства за лечение на туморни клетки на мозъка и бъбреците. В контекста на поведенческите проблеми е необходимо специфично лечение на детето.
Лечението на заболяването обикновено е дългосрочно. (1)